
Por Brian Miller
Um dos pilares centrais do modelo evolucionário padrão é a crença de que todas as espécies vivas evoluíram de um ancestral comum aparentando uma árvore da vida que se desdobra gradualmente. Como resultado, acredita-se que o padrão de semelhanças e diferenças nas espécies se encaixa em um padrão de árvore ou hierarquia aninhada, onde os pontos de ramificação correspondem ao aparecimento de novas características. Por exemplo, todos os mamíferos compartilham certas características, como a produção de leite, uma vez que seu ancestral comum mais recente desenvolveu essas características, e as características foram herdadas em cada ramo em desenvolvimento da árvore. Esta árvore da vida icônica foi apresentada ao público como uma das mais fortes evidências para o desenvolvimento da vida, progredindo inteiramente através de processos naturais não direcionados. No entanto, muitos aspectos desta história foram contraditados por grandes descobertas ao longo das últimas décadas.
De primordial importância, os primeiros representantes da maioria dos principais grupos de organismos apareceram repentinamente no registro fóssil, sem sequências identificáveis de intermediários que remontam a um ancestral comum com outros grupos. Igualmente problemático, o padrão de características físicas e sequências moleculares em espécies ao longo da natureza não implica em uma árvore evolucionária consistente. Por exemplo, os olhos dos humanos e dos polvos são bem parecidos, mas acredita-se que os dois grupos estejam apenas distantemente relacionados. Destacando este desafio, um artigo mais antigo analisou em vários estudos as porcentagens de características para determinados grupos de espécies que se encaixam consistentemente com os cladogramas melhor construídos (aproximações para uma árvore evolucionária). Essas porcentagens conhecidas como “índices de consistência” foram então plotadas no mesmo gráfico que as derivadas de dados gerados aleatoriamente, e os índices foram então ajustados para remover o efeito do ruído aleatório. A média dos índices ajustados residia em torno de 0,35. Tentativas mais recentes de construir cladogramas para vários grupos não se saíram melhor, como com Euarchonta (grupo incluindo primatas) e Therapsids (antepassados propostos a mamíferos). Em outras palavras, aproximadamente dois terços de todos os dados não se encaixam no modelo de ancestralidade comum.
Resultados decepcionantes, mecanismos ad hoc
Esses resultados decepcionantes exigiram que os evolucionistas inventassem vários mecanismos ad hoc para explicar as incoerências onipresentes. Exemplos incluem Transferência Horizontal de Genes (THG), perda diferencial de genes e evolução convergente. No entanto, o apelo recorrente à THG tem sido seriamente questionado. E a alegação de que adaptações complexas podem aparecer de forma independente várias vezes (evolução convergente) entra em colapso em um exame minucioso devido à implausibilidade de sua aparição por meio de processos não direcionados, mesmo uma vez.
Por exemplo, acredita-se que os olhos com lentes tenham evoluído de forma independente várias vezes, mas todos os cenários enfrentam barreiras intransponíveis em termos de pressões seletivas opostas e escalas de tempo necessárias. Ainda mais impressionante, a suposta evolução convergente da ecolocalização em morcegos e golfinhos envolveu as mesmas modificações de sequência em mais de 200 regiões do seu DNA. No entanto, o tempo disponível para um mamífero terrestre evoluir para um animal marinho totalmente aquático é insuficiente para adquirir até duas novas mutações coordenadas, e a transformação em mamíferos aquáticos exigiu muitas modificações adicionais. Apesar desses desafios, os evolucionistas afirmam que a ancestralidade comum ainda é a melhor explicação para os dados, já que de certa forma se ajusta a um padrão semelhante a uma árvore. Essa afirmação agora enfrenta um desafio formidável de um artigo recente na revista BIO-Complexity, de Winston Ewert. Ele persuasivamente apresenta uma nova estrutura para explicar melhor o padrão da natureza.¹
Nota do tradutor ¹: É o mesmo padrão que percebi e apresentei, de certa forma, em As Árvores Informacionais da Vida e Argumento de Coerência: Design versus Unidade do Design. Seria muito difícil esses padrões escaparem a especialistas em sistemas de informação.
Um novo framework
O modelo de Ewert interpreta o padrão de similaridades em diferentes grupos de espécies como se encaixando no que é referido na ciência da computação como gráficos de dependência. Especificamente, os programadores normalmente não escrevem novos programas inteiramente do zero. Em vez disso, eles reutilizam módulos padrão. Um exemplo é a solicitação do módulo JavaScript, que faz o download de arquivos da Internet. Os módulos geralmente acessam outros módulos, formando uma rede ramificada de relacionamentos de dependência (veja a Figura 1).
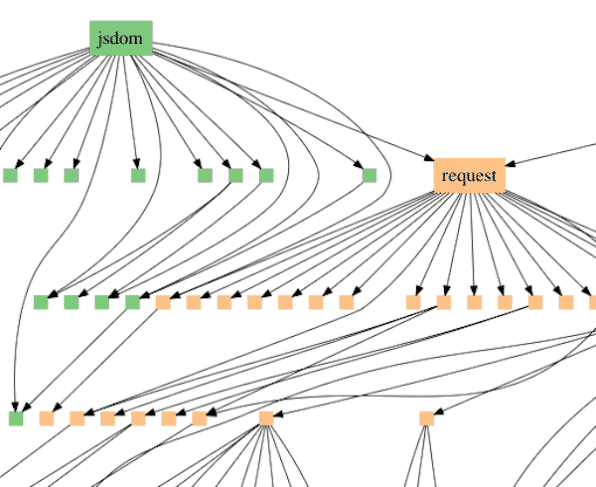
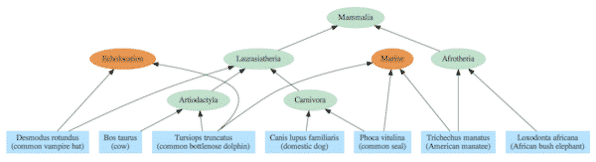
Os gráficos de dependência geralmente incluem uma hierarquia aninhada como parte de sua estrutura, mas também contêm relacionamentos adicionais que se estendem além de uma árvore simples. A diferença pode ser vista comparando a árvore de vida padrão dos mamíferos (Figura 2) com base na descendência comum com um gráfico de dependência derivado da mesma espécie (Figura 3).
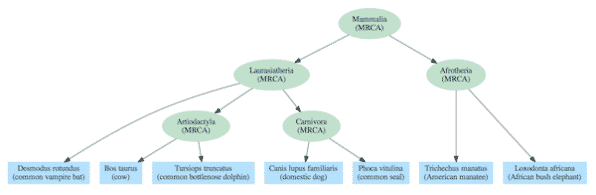
O modelo de árvore evolutiva requer, pelo menos para a vida complexa, que todas as espécies se liguem a um ancestral comum mais recente (ACMR) com outras espécies do mesmo grupo (clade). Por exemplo, elefantes e peixes-boi são ambos membros do clado Afrotheria, então eles são descritos como tendo um ACMR que foi o primeiro representante desse clade. E cada ACMR conecta-se a um único ACMR para todos os membros dos clados em níveis mais altos na hierarquia. Por exemplo, os ACMRs dos clados Artiodactyla e Carnivora compartilham um único ACMR com todos os membros do clado Laurasiatheria. E todas as espécies de mamíferos se ligam a um único ACMR que é o tronco da árvore da vida dos Mammalia.
O gráfico de dependência correspondente inclui a mesma hierarquia aninhada que a árvore evolucionária suposta, mas os relacionamentos são interpretados não em termos de ACMRs, mas em termos de módulos compartilhados. Por exemplo, elefantes e peixes-boi usam o módulo Afrotheria e todos os mamíferos usam o módulo Mammalia. No entanto, em contraste com o modelo de ancestralidade comum, peixes-boi, focas e golfinhos usam o módulo marinho que é incongruente com a árvore evolucionária. Da mesma forma, morcegos e golfinhos usam o módulo de ecolocalização, enquanto no modelo de ancestralidade comum eles não estão intimamente relacionados.
Comparando Dois Modelos
Ewert comparou o poder preditivo do gráfico de dependência e modelos de ancestralidade comum analisando a distribuição de famílias de genes em diferentes coleções de espécies retiradas de nove bancos de dados diferentes. Para cada base de dados, um conjunto de genes usados por várias espécies foi identificado como um módulo do qual essas espécies dependem. E um conjunto de genes contidos em vários módulos maiores foi identificado como um módulo distinto do qual os módulos maiores dependem. Uma rotina de otimização foi usada para construir uma aproximação para o melhor gráfico de dependência, e esse gráfico foi comparado à árvore da vida como apresentado pela hierarquia do banco de dados do NCBI. O melhor ajuste para os dados foi então determinado entre as duas representações usando o modelo de seleção bayesiano.
O modelo de gráfico de dependência faz várias previsões que estão em oposição direta ao modelo de ancestralidade comum:
- Dados biológicos devem ajustar um gráfico de dependência melhor que uma árvore.
- Dados produzidos por um processo dominado por descendência comum ou ramificação devem se ajustar a uma árvore melhor do que um gráfico de dependência.
- Gráficos inferidos de dados biológicos devem conter muito mais módulos não-taxonômicos com muito mais genes do que gráficos de dependência inferidos de dados que se sabe terem sido produzidos por descendentes comuns.
- O software deve ajustar a um gráfico de dependência melhor do que a uma árvore, mas uma árvore melhor do que um modelo nulo. Um modelo nulo corresponde a nenhum padrão existente para o reuso de famílias de genes entre as espécies.
A análise de Ewert validou todas essas previsões com alta confiança estatística para todos os bancos de dados. Portanto, este estudo inicial sugere que o modelo de gráfico de dependência supera em muito o modelo de ancestralidade comum para entender o padrão da natureza.
Como consequência, todas as supostas árvores e sequências evolutivas tornam-se altamente suspeitas, incluindo ícones como as séries de baleias e humanas. Pois eles são baseados em similaridades de características entre espécies, e similaridades são um indicador não confiável de ancestralidade comum, como está implícito nos índices de consistência tipicamente baixos das árvores. Em vez disso, as semelhanças parecem ser o resultado do uso de módulos de design comuns em diferentes espécies para atingir objetivos comuns.
O artigo de Ewert representa apenas o primeiro passo para avaliar e desenvolver seu framework. Ainda assim, o significado desta pesquisa não pode ser extrapolado. O modelo de gráfico de dependência explica por que os subconjuntos dos dados biológicos se encaixam grosseiramente em um padrão de árvore e por que muitos dos dados são incongruentes. Ele também faz previsões claras sobre os resultados de estudos futuros sobre a distribuição entre espécies de características físicas e semelhanças em dados moleculares. Finalmente, deve levar a um programa de pesquisa robusto e inovador, baseado em um framework do Design Inteligente².
Nota do tradutor ²: Como eu disse em As Árvores Informacionais da Vida, infelizmente esse modelo é um tanto mais difícil de ser falseado, seria uma espécie de “seleção natural” da nossa teoria. O importante é que seja verdadeiro.
Brian Miller. BIO-Complexity Presents Better Model than Common Ancestry for Explaining Pattern of Nature. July 19, 2018, 1:30 AM.
(Acessar)
Faça um comentário