
É comum na engenharia e no desenvolvimento de elementos funcionais a noção de que “a forma segue a função”. Isso porque a função depende muito da forma. Na biologia não é diferente, por essa razão o design tem se tornado um princípio unificador. As proteínas – que respondem pelas atividades metabólicas, controle, sinalização e outros processos em todos os subsistemas da vida – dependem da conformação assumida para desempenharem seus papéis. A conformação das proteínas depende da ordem e propriedades das subunidades que a compõe, os aminoácidos.
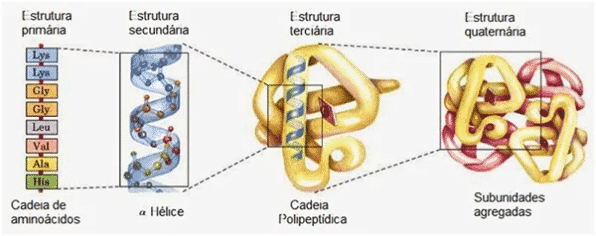
Considerando o número de aminoácidos e as sequências possíveis que eles podem formar com o tamanho médio de proteínas (~466 aminoácidos), o problema de combinatória se tornou claro: como variações aleatórias alcançariam funcionalidades em um universo de possibilidades tão grande? Em 1966, no Wistar Institute, matemáticos e cientistas se reuniram para avaliar a viabilidade deste cenário, o resultado foi a obra “Desafios matemáticos para a interpretação neodarwinista da evolução”, em que, apesar de inconclusiva pelas limitações da época, a conclusão foi desfavorável ao neodarwinismo:
"Nenhuma linguagem formal existente hoje pode tolerar mudanças aleatórias nas sequências de símbolos que expressam suas sentenças. O significado é quase invariavelmente destruído." Murray Eden. Wistar Institute, Filadélfia, 1966
Duas ideias foram consideradas por conta da perspectiva de origem evolutiva: as sequências de aminoácidos eram aleatórias (1) e um grande número de sequências alcançam conformação funcional (2).
A primeira ideia foi refutada por Sanger ao mapear a sequência da insulina. A segunda ideia foi refutada por Douglas Axe através de mutagénese sítio-específica, uma técnica que produz um grande número de variações de sequências, foi possível avaliar o número de sequências funcionais em um espaço de possibilidades.
Para se ter uma ideia uma proteína com 150 aminoácidos, considerada pequena, pode assumir 10 elevado à 195ª potência de possíveis arranjos. Os cientistas não sabiam quantas seriam funcionais para avaliar a possibilidade de caminhos evolutivos. Muitos continuavam sustentando que as sequências funcionais não eram tão raras. Mas todas essas especulações foram substituídas com dados experimentais.
A partir de uma proteína funcional de 150 aminoácidos, Axe demonstrou que as sequências funcionais são, aproximadamente, apenas uma em cada 10 elevado à 77ª potência das sequências possíveis. Considere que existem apenas 10 elevado à 65ª átomos em nossa galáxia. Mas como relacionamos isso aos organismos vivos e a variação de proteínas?
O número de “tentativas” assume a forma de organismos vivos individuais nos quais as mutações ocorrem. Podemos estimar que o número de organismos que já existiram na Terra está por volta de 10 elevado à 40ª. Isso significa que todos os organismos que já existiram não alcançariam sequer uma única proteína funcional. E mesmo um pequeno organismo requer centenas de proteínas funcionais.
Axe sustenta que as proteínas estão em “ilhas de funcionalidades”. Isso significa que ligeiras variações ainda podem possuir função (inclusas nas estimativas), mas as drásticas transformações exigidas em histórias evolutivas são refutadas categoricamente. De fato, as proteínas possuem um ajuste de tolerância à mutações e o próprio código genético conta com uma espécie de “código Hamming” interno para minimização de erros universal.
Atualmente vários projetos rodam usando computadores com centenas de milhares de unidades de processamento para aproximação de dobras de proteínas – as dobras que garantem a funcionalidade. Na página de um desses projetos, o Folding@home, você pode ver algumas estatísticas. Entender como as proteínas adotam sua estrutura tridimensional continua sendo um dos maiores desafios da ciência. O grande consumo da capacidade de processamento nos apresenta uma “engenharia da exaustão”.
As taxas de mutações em todos os organismos que já existiram juntos não seriam suficientes para encontrar sequências funcionais. Ferramentas de edição de genoma como a CRISPR/Cas são adaptações ou cópias de sistemas encontrados em bactérias. É praticamente inviável hoje desenvolver qualquer proteína original sem se basear nos dados de proteínas conhecidas e por isso nossa habilidade de inovação na engenharia biológica é limitada ao que descobrimos, diferente das outras engenharias onde temos mais facilidade de criação.
Agora vem a pergunta: se nós, com toda nossa capacidade atual de processamento, temos dificuldade com isso, e se a inferência probabilística é extremamente contrária, por que ainda sustentam as crenças de mutações aleatórias e seleção natural como responsáveis pelo design biológico?
Referências
Axe, Douglas D. “Estimating the prevalence of protein sequences adopting functional enzyme folds.” Journal of Molecular Biology 341.5 (2004): 1295-1315.
Moorhead, Paul S. “Mathematical challenges to the neo-Darwinian interpretation of evolution; a symposium held at the Wistar Institute of Anatomy and Biology April 25 and 26, 1966.” (1967).
Wei, Leyi, and Quan Zou. “Recent progress in machine learning-based methods for protein fold recognition.” International journal of molecular sciences 17.12 (2016): 2118.
…
Faça um comentário