Nota do tradutor: esta é a parte 6 da série de 10 artigos sobre os problemas científicos da evolução biológica e química. A série é baseada no capítulo “The Top Ten Scientific Problems with Biological and Chemical Evolution” de autoria de Casey Luskin no livro More than Myth, editado por Paul Brown e Robert Stackpole (Chartwell Press, 2014). Eis a lista de todos os artigos da série: Artigo introdutório, Problema 1, Problema 2, Problema 3, Problema 4, Problema 5, Problema 6, Problema 7, Problema 8, Problema 9, Problema 10.
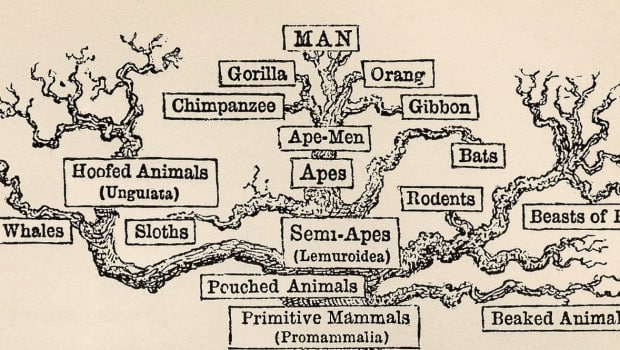
Quando fósseis não conseguem demonstrar que os animais evoluíram de um ancestral comum, os cientistas evolucionistas viraram-se para um outro tipo de evidência – os dados sequenciais do DNA – para demonstrar uma “árvore da vida”. Na década de 1960, no tempo em que o código genético foi compreendido pela primeira vez, os bioquímicos Émile Zuckerkandl e Linus Pauling montaram a hipótese de que, se as sequências de DNA poderiam ser usadas para produzir árvores evolutivas – árvores que combinavam com aquelas que eram baseadas em características morfológicas ou anatômicas – então isso iria fornecer “a melhor e singela prova disponível da realidade da macroevolução” [99]. Assim então começou-se um esforço de décadas para sequenciar os genes de vários organismos e para construir árvores “moleculares” com base evolutiva (“filogenéticas”). O objetivo final é o de construir um grande “árvore da vida”, mostrando a forma como todos os organismos vivos estão relacionados através da ancestralidade comum universal.
A premissa principal
A lógica básica por trás de construir árvores moleculares é relativamente simples. Em primeiro lugar, os investigadores escolhem um gene, ou um conjunto de genes, encontrados em vários organismos. Em seguida, os genes são analisados para determinar suas sequências de nucleotídeos, de modo que as sequências de genes de vários organismos possam então ser comparadas. Finalmente, uma árvore evolutiva é construída com base no princípio de que quanto mais semelhante for a sequência de nucleotídeos, mais estreitamente relacionadas são as espécies. Um artigo no periódico Biological Theory assim fala:
A Sistemática Molecular é (em grande parte) baseada na suposição, articulada pela primeira vez por Zuckerkandl e Pauling (1962) de maneira clara, de que o grau de semelhança global reflete o grau de parentesco. [100]
Essa suposição é, em essência, uma articulação de uma das maiores características da teoria – a idéia de ancestralidade comum universal. No entanto, é importante perceber que é uma mera suposição reivindicar que as semelhanças genéticas entre espécies diferentes sejam resultado necessário de uma ancestralidade comum.
Pensando dentro de um paradigma estritamente darwinista, esses pressupostos fluem naturalmente. Assim como o já mencionado artigo da Biological Theory explica, a principal premissa subjacente às árvores moleculares “são deduzidas de interpretações de similaridade molecular (ou dissimilaridade) entre táxons no contexto do modelo darwiniano de mudança contínua e gradual” [101]. Então, já se assume que a teoria seja verdadeira para construir a árvore. Mas também, se a evolução darwiniana fosse verdadeira, a construção de árvores usando sequências diferentes deveria revelar um padrão razoavelmente consistente nos diferentes genes ou sequências.
Isso torna ainda mais significativo o fato de que os esforços para construir uma grande “árvore da vida” usando DNA ou outros dados de seqüência biológica não se enquadram nas expectativas. O problema básico é que um gene dá uma versão da árvore da vida, enquanto outro gene dá uma versão muito diferente e conflitante da árvore. Por exemplo, como vamos discutir mais adiante, a árvore padrão dos mamíferos coloca os seres humanos mais proximamente relacionados com os roedores do que os elefantes. Mas os estudos de um determinado tipo de DNA chamado genes de microRNA têm sugerido o contrário – que os seres humanos eram mais próximos aos elefantes do que ao roedores. Tais conflitos entre árvores baseados em genes são bastante comuns.
Os dados genéticos, portanto, não estão formando um quadro consistente de ancestralidade comum, mostrando que as premissas por trás da construção das árvores falham comumente. Isto leva a perguntas justificáveis sobre a ancestralidade comum universal estar correta ou não.
Conflitos na base da árvore da vida
Os primeiros problemas surgiram quando os biólogos moleculares sequenciaram genes a partir dos três domínios básicos da vida – bactérias, procariontes e eucariontes – mas esses genes não permitiam que esses grupos básicos de vida pudessem ser resolvidos em um padrão parecido com árvore. Em 2009, a revista New Scientist publicou uma reportagem de capa intitulada, “Por que Darwin estava errado sobre a árvore da vida”, que explicava esses dilemas:
Os problemas começaram no início da década de 90, quando se tornou possível sequenciar genes reais de bactérias e procariontes, em vez de apenas RNA. Todos esperavam que essas sequências de DNA confirmassem a árvore de RNA, e em algumas vezes elas confirmavam mas, fundamentalmente noutras vezes não. O RNA, por exemplo, poderia sugerir que a espécie A foi mais intimamente relacionada com a espécie B do que com a espécie C, mas uma árvore construída a partir do DNA sugeriria o inverso. [102]
Esse tipo de dado levou o bioquímico W. Ford Doolittle a explicar que “os filogenistas moleculares terão falhado em encontrar a ‘árvore verdadeira’, não porque seus métodos sejam inadequados ou porque eles escolheram os genes errados, mas porque a história da vida não pode ser representada apropriadamente como uma árvore” [103]. A New Scientist assim coloca: “Por muito tempo, o santo graal foi construir a árvore da vida (…) Mas hoje o projeto está em farrapos, rasgado em pedaços por um violento ataque de provas negativas” [104].
Muitos evolucionistas respondem algumas vezes que esses problemas só surgem quando se estudam microrganismos como bactérias – organismos que podem trocar genes através de um processo chamado de “transferência horizontal de genes”, assim confundindo o sinal das relações evolutivas. Mas essa objeção não é tão verdadeira, dado que a árvore da vida é desafiada até mesmo entre os organismos superiores, onde tal troca de genes não é prevalente. Carl Woese, um pioneiro da sistemática molecular evolucionista, explica:
As incongruências filogenéticas podem ser vistas em toda parte da árvore universal, desde sua raiz até as principais ramificações dentro e entre os vários táxons até a composição dos próprios grupos primários [105].
Da mesma forma, o artigo da New Scientist observa que “pesquisas sugerem que a evolução de animais e plantas também não seja exatamente como uma árvore” [106]. O artigo explica o que aconteceu quando o microbiologista Michael Syvanen tentou criar uma árvore que mostra as relações evolutivas usando 2000 genes a partir de um grupo diverso de animais:
Ele falhou. O problema era que os diferentes genes contavam histórias evolutivas contraditórias (...) Os genes estavam dando sinais mistos (...) Cerca de 50 por cento dos seus genes têm uma história evolutiva, e 50 por cento, outra. [107]
Os dados estavam tão difíceis de serem solucionados numa árvore que Syvanen lamentou: “Nós simplesmente aniquilamos a árvore da vida” [108]. Muitos outros trabalhos na literatura técnica reconhecem problemas semelhantes.
Conflitos nos ramos mais altos
Um artigo de 2009 no periódico Trends in Ecology and Evolution observa que “um grande desafio para incorporar essas grandes quantidades de dados numa inferência de árvore das espécies é que muitas histórias genealógicas conflitantes existem em diferentes genes por todo o genoma” [109]. De jeito similar, um artigo na Genome Research estudou as sequências de DNA em vários grupos de animais e descobriu que “diferentes proteínas geram diferentes árvores filogenéticas” [110]. Um artigo de junho de 2012 na revista Nature informou que cadeias curtas de RNA chamado microRNAs “estão destruindo idéias tradicionais sobre a árvore genealógica dos animais”. O biólogo de Dartmouth Kevin Peterson, que estuda microRNAs lamentou: “Eu olhei para milhares de genes de microRNA, e eu não consigo encontrar um único exemplo que apoiasse a árvore tradicional”. De acordo com o artigo, os microRNAs produziram “um diagrama radicalmente diferente para os mamíferos: um que coloca os seres humanos mais próximos dos elefantes do que com os roedores”. Peterson diz sem rodeios: “Os microRNAs são totalmente inequívocos… eles dão uma árvore totalmente diferente do que todo mundo quer” [111].
Conflitos entre moléculas e morfologia
Nem todas as árvores filogenéticas são construídas pela comparação de moléculas como o DNA de espécies diferentes. Muitas árvores são baseadas em comparação da forma, da estrutura e do plano corpóreo de organismos diferentes – também chamada de “morfologia”. Mas conflitos entre árvores baseadas no estudo de moléculas e árvores baseadas na morfologia também são comuns. Um artigo de 2012 que estudou as relações entre morcegos deixou isso claro, afirmando: “Incongruências entre filogenias obtidas das análises moleculares e das morfológicas, e entre as árvores baseadas em diferentes subconjuntos de sequências moleculares tornaram-se generalizadas, enquanto os conjuntos de dados têm se expandido tanto em características e espécies” [112]. Dificilmente esse é o único estudo a encontrar conflitos entre árvores baseadas em DNA e árvores baseadas em características anatômicas ou morfológicas. Livros didáticos muitas vezes defendem a ancestralidade comum usando o exemplo de uma árvore de animais baseada na enzima citocromo c, que corresponde à árvore evolutiva tradicional baseada na morfologia [113]. No entanto, os livros didáticos raramente mencionam que a árvore baseada em uma enzima diferente, citocromo b, entra em conflito acentuado com a árvore evolutiva padrão. Um artigo na Trends in Ecology and Evolution observou:
O gene do citocromo b mitocondrial sugeriu [...] uma filogenia absurda de mamíferos, independentemente do método de construção da árvore. Gatos e baleias caíram entre primatas, agrupando-se com símios (macacos e orangotangos) e estrepsirrinos (lêmures, gálagos e lorisídeos). O citocromo b é provavelmente o gene mais comumente seqüenciado dos vertebrados, o que faz deste resultado surpreendente ainda mais desconcertante. [114]
Surpreendentemente, um artigo diferente em Trends in Ecology and Evolution concluiu, “a riqueza das propostas morfológicas concorrentes, bem como as propostas moleculares das filogenias predominantes das ordens dos mamíferos, reduziria a árvore dos mamíferos para um arbusto sem solução, sendo a única relação evolutiva consistente sendo, provavelmente, o agrupamento de elefantes e sirênios” [115]. Por causa desses conflitos, um importante artigo de revisão na revista Nature relatou, “as disparidades entre as árvores moleculares e morfológicas” levaram a “guerras da evolução”, porque “as árvores evolutivas construídas pelo estudo de moléculas biológicas muitas vezes não se assemelham às elaboradas a partir da morfologia” [116].
Finalmente, um estudo publicado na revista Science, em 2005, tentou usar genes para reconstruir as relações do filos animais, mas concluiu que “apesar da quantidade de dados e da largura de táxons analisados, as relações entre a maioria dos filos [de animais] permanecem sem solução”. No ano seguinte, os mesmos autores publicaram um artigo científico intitulado “Bushes in the Tree of Life”, que ofereceu conclusões impressionantes. Os autores reconhecem que “uma grande fração de genes individuais produzem filogenias de má qualidade”, observando que um estudo “omitiu 35% dos genes individuais de sua matriz de dados, porque esses genes produziram filogenias em desacordo com a sabedoria convencional”. O artigo sugere que “certas partes críticas da árvore da vida podem ser difíceis de resolver, independentemente da quantidade de dados convencionais disponíveis”. O artigo ainda afirma que “as recorrentes descobertas de clades persistentemente sem solução (arbustos) devem forçar uma reavaliação de várias suposições amplamente sustentadas de sistemática molecular” [117].
Infelizmente, um pressuposto que esses biólogos evolucionistas não estão dispostos a reavaliar é o pressuposto de que a ancestralidade comum universal esteja correta. Eles apelam a uma miríade de argumentos ad hoc – da transferência horizontal de genes, atração de ramos longos, evolução rápida, diferentes taxas de evolução, teoria coalescente, amostragem incompleta, metodologia falha, evolução convergente – para explicar dados inconvenientes que não se encaixam no padrão de árvore desejado. Como um artigo de 2012 declarou: “o conflito filogenético é comum e é frequentemente a norma, e não a exceção” [118]. No final do dia, o sonho de que os dados da sequência do DNA se encaixariam em uma fina e ajustada árvore de vida falhou, e com ele, uma previsão fundamental da teoria neo-darwinista.
Texto traduzido e adaptado de Evolution News & Views.
Referências:
[99] Zuckerkandl and Pauling, “Evolutionary Divergence and Convergence in Proteins”, 101.
[100] Jeffrey H. Schwartz, Bruno Maresca, “Do Molecular Clocks Run at All? A Critique of Molecular Systematics”, Biological Theory, 1(4):357-371, (2006).
[101] Ibid.
[102] Graham Lawton, “Why Darwin was wrong about the tree of life”, New Scientist (21 de janeiro de 2009).
[103] W. Ford Doolittle, “Phylogenetic Classification and the Universal Tree”, Science, 284:2124-2128 (25 de junho de 1999).
[104] Citando parcialmente Eric Bapteste, em Lawton, “Why Darwin was wrong about the tree of life” (citações internas omitidas).
[105] Carl Woese, “The Universal Ancestor”, Proceedings of the National Academy of Sciences USA, 95:6854-9859 (junho de 1998) (ênfase adicionada).
[106] Graham Lawton, “Why Darwin was wrong about the tree of life”, New Scientist (21 de janeiro de 2009).
[107] Citando parcialmente Michael Syvanen, emLawton, “Why Darwin was wrong about the tree of life” (citações internas omitidas).
[108] Michael Syvanen, citado em Lawton, “Why Darwin was wrong about the tree of life”.
[109] James H. Degnan e Noah A. Rosenberg, “Gene tree discordance, phylogenetic inference and the multispecies coalescent”, Trends in Ecology and Evolution, 24 (2009): 332-340.
[110] Arcady R. Mushegian, James R. Garey, Jason Martin e Leo X. Liu, “Large-Scale Taxonomic Profiling of Eukaryotic Model Organisms: A Comparison of Orthologous Proteins Encoded by the Human, Fly, Nematode, and Yeast Genomes”, Genome Research, 8 (1998): 590-598.
[111] Elie Dolgin, “Rewriting Evolution”, Nature, 486: 460-462 (June 28, 2012).
[112] Liliana M. Dávalos, Andrea L. Cirranello, Jonathan H. Geisler, e Nancy B. Simmons, “Understanding phylogenetic incongruence: lessons from phyllostomid bats”, Biological Reviews of the Cambridge Philosophical Society, 87:991-1024 (2012).
[113] Por exemplo, ver BSCS Biology: A Molecular Approach (Glencoe/McGraw Hill, 2006), 227; Sylvia S. Mader, Jeffrey A. Isaacson, Kimberly G. Lyle-Ippolito, Andrew T. Storfer, Inquiry Into Life, 13ª ed. (McGraw Hill, 2011), 550.
[114] Ver Michael S. Y. Lee, “Molecular Phylogenies Become Functional”, Trends in Ecology and Evolution, 14: 177 (1999).
[115] W. W. De Jong, “Molecules remodel the mammalian tree”, Trends in Ecology and Evolution, 13(7), pp. 270-274 (7 de julho de 1998).
[116] Trisha Gura, “Bones, Molecules or Both?”, Nature, 406 (20 de julho de 2000): 230-233.
[117] Antonis Rokas & Sean B. Carroll, “Bushes in the Tree of Life,” PLoS Biology, 4(11): 1899-1904 (novembro de 2006) (citações internas e figuras omitidas).
[118] Liliana M. Dávalos, Andrea L. Cirranello, Jonathan H. Geisler e Nancy B. Simmons, “Understanding phylogenetic incongruence: lessons from phyllostomid bats”, Biological Reviews of the Cambridge Philosophical Society, 87:991-1024 (2012).
Faça um comentário